Challenges in Vaccine Design
The short version: Here is a very nerdy post on why it’s hard to make an effective vaccine. HIV vaccines are a fantastic example of the challenges because basically everything that could be a challenge in making a vaccine in theory is a challenge in HIV vaccine design. In general, it’s hard to make vaccines when you have many strains of a pathogen, it’s hard when you don’t know exactly what the immune system needs to do to protect against disease (this is actually very complicated- it’s not all antibodies), it’s very hard to do when you are trying to vaccinate the very young and the very not, and it can be very hard to do if you need good protection at mucosal surfaces (like the respiratory, gastrointestinal, and genitourinary tract). In short: it’s hard.
Recently I’ve gotten a bunch of questions that more or less center around why vaccinology is hard. Why will it take so long for us to have a COVID-19 vaccine? Are there unique challenges with making a vaccine for the elderly and for the very young? Why do we sometimes fail to respond to vaccines? All this and more covered in this post. Feel free to jump to the section that you are most interested in (I appreciate this is long).
The Core Challenges of Vaccination
Source: Plotkin’s Vaccines 7th Edition Table 29.1
The reality is that all of the easily won vaccines probably already exist. There are a few reasons why it’s so hard to make a vaccine, and the archetype of a challenge for vaccination is best exemplified with HIV, and some of the reasons are discussed in the table below.
I think these are worth elaborating upon and discussing in the context of other diseases as well.
Correlates of Protection
Often, I hear it cited that people think that vaccine-induced immunity is inferior to disease-induced immunity because it is “unnatural.” The reality is however, vaccines are designed with the intention of mirroring the infection as closely as possible to elicit the appropriate correlates of protection. Correlates of protection are variables that can be measured in the course of an immune response which can be used to determine whether or not an individual is protected against disease. They are split into mechanistic and nonmechanistic correlates of protection. Mechanistic correlates of protection are those variables which are themselves responsible for protection. For example, with measles-containing vaccines the mechanistic correlates of protection are the memory CD8 T cells which will kill any virally infected cells and the anti-peplomer (peplomers are the surface glycoproteins that mediate attachment and entry into cells) neutralizing antibodies that prevent viral entry into the cells. On the other hand, with nonmechanistic correlates of protection, the variable being measured is highly correlated with the actual mechanistic correlate of protection but not itself doing the protecting. For example, with varicella (chickenpox and shingles) memory CD8 T cells are the most important protective measure in ensuring that reactivation of latent virus (shingles) does not occur (the mechanistic correlate). However, these are highly correlated with anti-varicella antibodies, and thus we rely on antibody titers to determine whether or not someone is protected. CD8 T cells can also be measured to determine whether someone is protected, but it is a considerably more involved and expensive process. Determining the appropriate correlates of protection however, comes from observing those who recover from infection and seeing what type of immune response they mount to clear the offending pathogen. Vaccines are then produced with antigens and adjuvants (if necessary) to elicit those same kinds of responses. In some cases (elaborated upon below) this is not always possible, either because it would be unsafe or because our current technology has not advanced to such a point. Hence we might go for second-best or third-best knowing that some protection is always going to be better than none. In addition, an important subtlety that is often missed in discussing correlates of protection is that they are defined with respect to a particular phenomenon within a disease, rather than the disease itself. For example, the quantity of anti-pneumoccocal antibody required to protect oneself against pneumococcal bacteremia (the spread of the bacteria throughout the blood) is considerably lower than what would be needed to prevent ear infections.
This is a key challenge with any vaccine candidate for COVID-19: we do not understand presently what the most important correlates of protection are. We are measuring antibodies in individuals and we do have evidence that most people will develop antibodies following infection (seroconversion). However, it is thus far unclear what the relative importance of these antibodies are with respect to recovery and prevention of disease, though this study suggests they will persist for at least 2 years. The role of T cell responses in the disease is also still not fully understood. For that reason it’s hard to know which correlates to target. The same is largely true of HIV and it’s actually worse because no one who has gotten HIV has ever spontaneously recovered- meaning that we don’t know what it takes for the immune system to get rid of HIV.
For most vaccines, the correlate of protection is an antibody against the vaccine antigen. Put another way, most vaccines protect via humoral immunity. Humoral immunity is an old term referencing the body’s humors (bodily fluids), in this case mainly plasma, and is one of the two major arms of the immune system. It generally refers to the protection conferred to by antibodies. Accomplishing this with our current understanding of vaccinology is not terribly difficult (though of course, not always), in particular through the aid of aluminum-based adjuvants. The other major arm of the immune system is cell-mediated immunity. Cell-mediated immunity is the use of cytotoxic T cells (aka CD8 T cells) specific to an antigen to kill infected cells. This however, is considerably more challenging to do and in general requires a live attenuated vaccine or a viral vector vaccine. This is a consequence of the actions of dendritic cells- the bridge between innate and adaptive immunity. Dendritic cells are specialized antigen presenting cells that are present initially in an immature form and then become activated upon encounter with antigen, which they then process and present to lymphocytes (see the previously linked aluminum-based adjuvants post for more information on that). Dendritic cells are special however because they are capable of a phenomenon known as cross-presentation and cross-priming (this gets a bit technical so bear with me):

Dendritic cell licensing for cross-presentation and CD8 T cell activation (cross-priming). Source: Kurts, C., Robinson, B. W. S., & Knolle, P. A. (2010). Cross-priming in health and disease. Nature Reviews Immunology, 10(6), 403–414. doi:10.1038/nri2780
The immune system works by responding to antigen, but it will not respond to any antigen- it will only respond to antigens properly presented. All cells except for red blood cells express a protein called major histocompatibility complex class I (MHC class I). MHC class I proteins are loaded with short fragments of antigen from the cytosol, antigens that originate from within the cell or endogenous antigen. On the other hand, antigen-presenting cells, in addition to MHC class I, also have MHC class II. MHC class II proteins are loaded with antigen that is taken up from the external environment (exogenous antigen). Now for the important point: helper T cells (CD4 T cells) recognize MHC Class II proteins, but CD8 T cells recognize only MHC Class I proteins. This is a problem in that it means CD8 T cells are not able to respond to any exogenously derived antigen (unless it is a cytosolic pathogen that infects dendritic cells). The solution however, already exists: cross-priming and cross-presentation (see the diagram to the immediate left). Dendritic cells are capable of breaking all the rules and displaying exogenous antigen on MHC class I proteins. This occurs in 2 steps. First the dendritic cell has to take up the antigen and receive “licensing” signals from a helper T cell (or in some cases, if there are alarmins present, this may be skipped and the dendritic cell can itself be licensed- but in general this is not the case because of cross-tolerance). Upon receiving these signals, the dendritic cell can now present exogenous antigen on its MHC Class I molecule, and supply the CD8 T cell with costimulatory signals needed for the induction of an immune response. The issue is: all vaccines represent exogenous antigens (at least, initially). They are not originating from within. The cytokines required to induce cross-presentation and cross-priming and overcome cross-tolerance are made by Th1 cells and this seems to require the use of TLR agonists as adjuvants (in particular, TLR9 agonists are good at this). Live attenuated viral vaccines are effective inducers of cross-presentation and cross-priming. For reasons that are not entirely clear, inactivated vaccines are not despite having all the same structural features (it would appear that replication of the antigen within the dendritic cell is important for effective cross-presentation to occur).
One of the factors that make mRNA vaccines such attractive candidates for COVID-19 is the fact that they are able to activate both arms of immunity at the same time (humoral and cell-mediated). Exogenous RNA is recognized by the immune system in a manner that a virus would be and activates pattern recognition receptors (PRRs) that can induce cross-presentation. Upon entry into the cytosol, the mRNA is transcribed and produces the spike protein, and in the inflammatory context provided by the mRNA’s own adjuvant effect, this can induce the formation of antibodies against the spike protein. However, this approach is not without drawbacks. Take it from someone who has worked with RNA before in an intimate manner: it’s hard. The thing will degrade if you look at it funny- by which I mean it is highly sensitive to temperature and pH changes, and thus difficult to store. Additionally, mRNA vaccines induce type 1 interferon, which while good inducers of inflammation necessary for an effective immune response and enhancers of cytotoxic T cell responses, make it more difficult for antigen to replicate (this can be remedied however by introducing noncanonical bases into the mRNA e.g. pseudouridine but at the cost of adjuvanticity). On the other hand, production is rapidly scalable and if this is the candidate that ends up working, we can meet the demand for a vaccine relatively quickly (compared with other approaches).
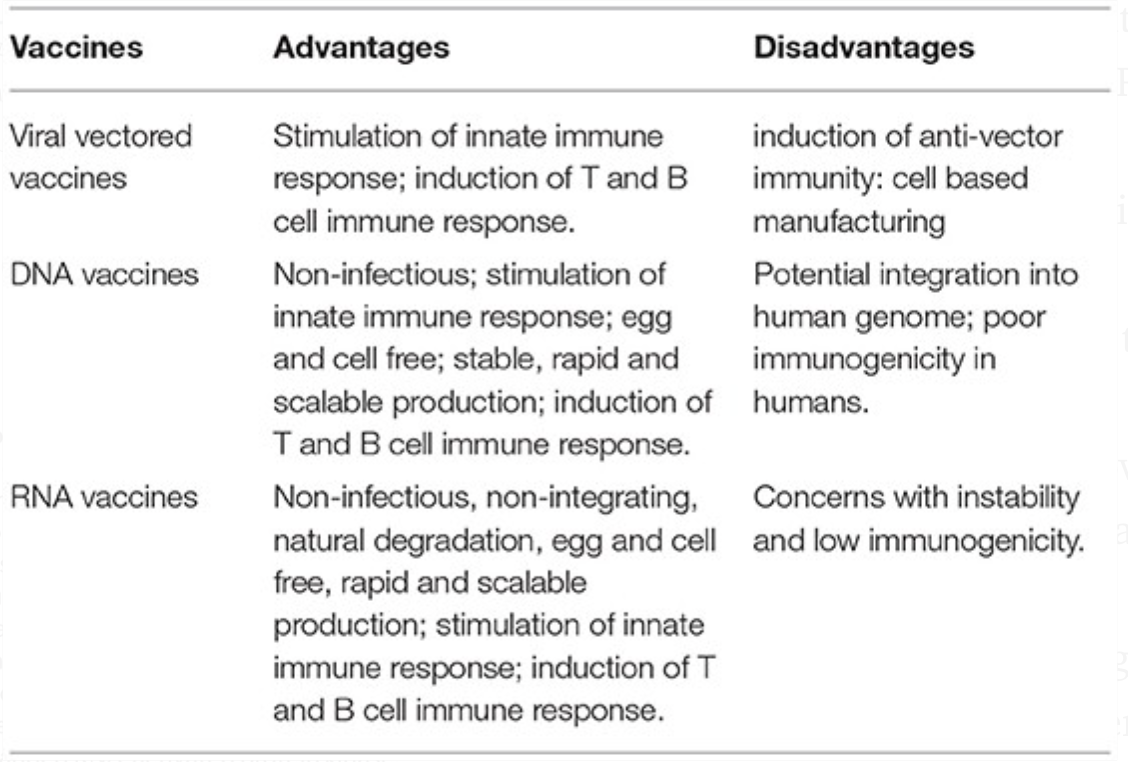
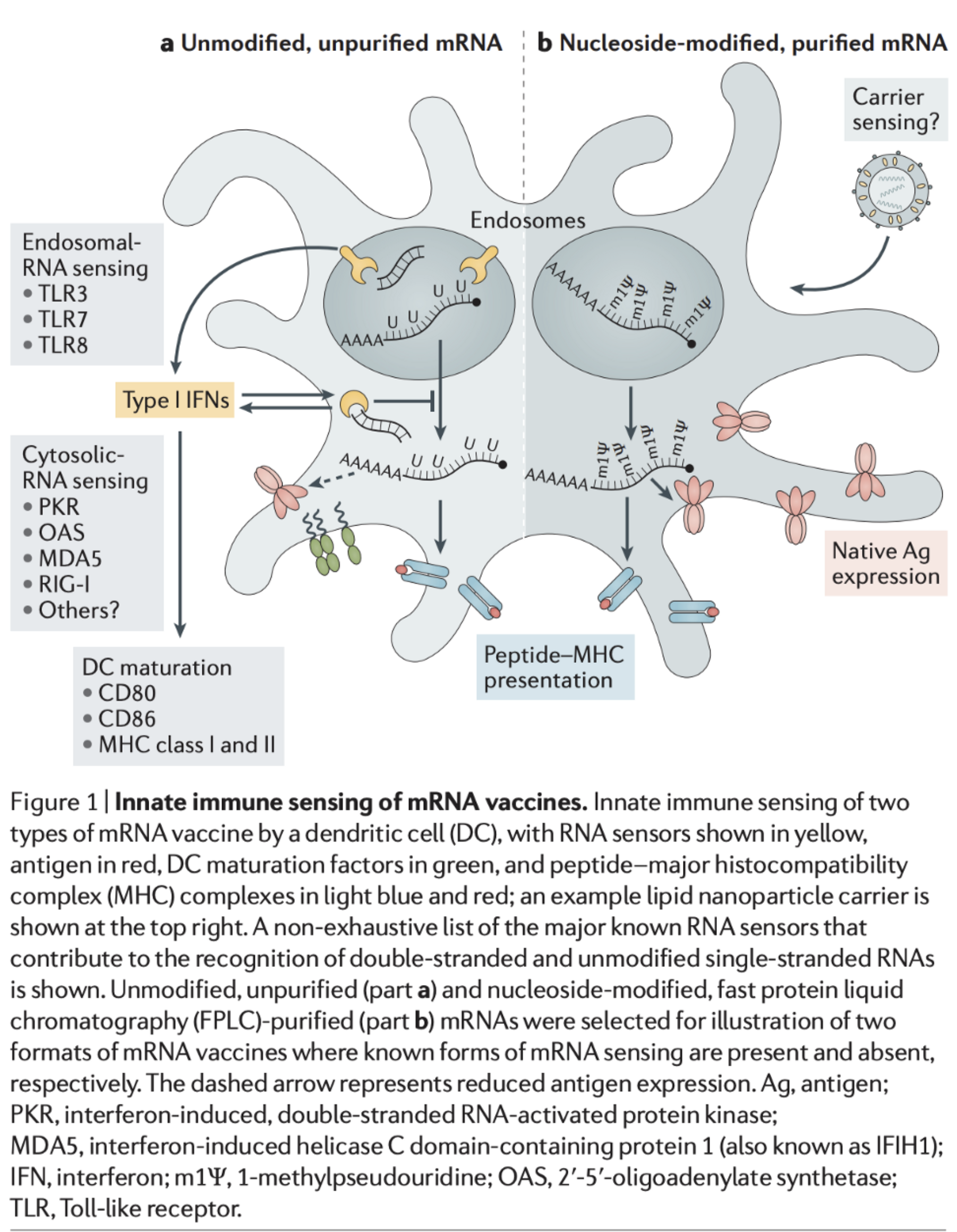
Source(s): Zhang C, Maruggi G, Shan H, Li J. Advances in mRNA Vaccines for Infectious Diseases. Frontiers in Immunology. 2019;10.
Pardi, N., Hogan, M., Porter, F. and Weissman, D., 2018. mRNA vaccines — a new era in vaccinology. Nature Reviews Drug Discovery, 17(4), pp.261-279.
One of the factors that make mRNA vaccines such attractive candidates for COVID-19 is the fact that they are able to activate both arms of immunity at the same time (humoral and cell-mediated). Exogenous RNA is recognized by the immune system in a manner that a virus would be and activates pattern recognition receptors (PRRs) that can induce cross-presentation. Upon entry into the cytosol, the mRNA is transcribed and produces the spike protein, and in the inflammatory context provided by the mRNA’s own adjuvant effect, this can induce the formation of antibodies against the spike protein. However, this approach is not without drawbacks. Take it from someone who has worked with RNA before in an intimate manner: it’s hard. The thing will degrade if you look at it funny- by which I mean it is highly sensitive to temperature and pH changes, and thus difficult to store. Additionally, mRNA vaccines induce type 1 interferon, which while good inducers of inflammation necessary for an effective immune response and enhancers of cytotoxic T cell responses, make it more difficult for antigen to replicate (this can be remedied however by introducing noncanonical bases into the mRNA e.g. pseudouridine but at the cost of adjuvanticity). On the other hand, production is rapidly scalable and if this is the candidate that ends up working, we can meet the demand for a vaccine relatively quickly (compared with other approaches).
Correlates of Vaccine-Induced Immunity. Source: Table 2.2 from Plotkin’s Vaccines 7th Edition
Antigenic Diversity
I’ll just say a brief aside on the antigenic diversity comment: this does not appear to be a concern with COVID-19 as, as of the time of writing this, there is no decent evidence for the existence of multiple strains of SARS-CoV-2. Yes, there are many discrete genotypic variants for SARS-CoV-2 but this does not a strain make. A strain has to have a meaningful set of characteristics that make it different from that of other strains e.g. a person who had an infection with one strain is protected against all pathogens of that strain but not of other strains (this is technically known as a serotype aka serovar). In this regard, though we see many genetic differences, there is still no convincing evidence for the presence of multiple strains. In particular, a recent preprint has described that SARS-CoV-2 from Europe has acquired a missense mutation in which an aspartic acid of the spike protein has been changed to a glycine at position 614 (hence D614G), and it speculates that this genotypic variant has increased virulence and communicability. However, I think my readers know at this point that correlation is not causation. In reality, this likely represents a founder effect. In essence, when a member of one population splits off and starts a new population, all new members of the population will have genetic resemblance to that original member. Unless there is an evolutionary selection pressure against it (that genotype has worse evolutionary fitness, e.g. it is worse at replicating), we expect the trait to persist unless it is accidentally removed. In other words, this paper basically shows that viruses that are derived from an ancestor in Europe resemble that European ancestor and claims about virulence and communicability are not justified based on the data they gathered- which is not to say they won’t be, but right now, the claims are not justified. See here for an excellent discussion by expert, Professor Angela Rasmussen.
This has, however, been a major challenge for other vaccines i.e. HIV and influenza. The remedy to this has been to seek out broadly neutralizing antibodies- antibodies that bind many different strains of the viruses, but thus far they have not borne fruitful vaccines. However, this has proved especially challenging in the case of HIV because the virus can modify its glycosylation patterns to conceal those broadly neutralizing epitopes.
Vaccination at the Extremes of Age
While not explicitly in Table 29.1, vaccinating the very young and very not young has always had challenges because of the way those two groups’ immune systems work. The outcomes of the immunological changes that these groups undergo are similar but stem from fundamentally different causes.
In the case of the very young, it is largely an outcome of pregnancy. During pregnancy, there are complex immunological changes that both the mother and the fetus undergo in the name of tolerance. The fetus represents a huge mass of non-self tissue, and while there is a placenta which serves as a barrier, the fetus can still release immunogenic material across it which would trigger a reaction in the mother. Hence, the mother undergoes complex immunologic changes to tolerate her fetus which had previously been referred to as immunosuppression but it’s now clear that this term is a bit misleading because pregnant women do not have a substantially higher incidence of infections. Similarly, the fetus’s immune system develops in utero as well and has to tolerate being intimately connected with a non-self environment. Recovery from this has a lag and thus the need for many doses of the same vaccine on the schedule, as infants are not very good at retaining immunological memory after they have been exposed to an antigen early in life. Additionally, there is concern for successive waves of the immune response disrupting the formation of productive immunological memory which is why vaccine appointments are always at least 3 weeks apart in the childhood schedule:
Source: Table 2.5 from Plotkin’s Vaccines 7th Edition
The effects on the immune system from infancy are apparent in virtually all aspects of immunologic function in infants. For example, neutrophils, a cell of the innate immune system, do not appear until very late in gestation and for some time the neutrophils have impaired phagocytic capacity and have difficulties with chemotaxis meaning that they have trouble taking up antigens like bacteria and getting to the site of inflammation. There are also reduced levels of pro-inflammatory cytokines made by dendritic cells and NK cells, further complicating things. In short, the reliable induction of a protective immune response in a neonate is challenging because their immune system is not primed to create conditions inhospitable for infection in the way that an adult’s is.
Source: Figure 2, Kollmann T, Marchant A, Way S. Vaccination strategies to enhance immunity in neonates. Science. 2020;368(6491):612–615.
Broadly speaking there are a few workarounds to make a neonate’s immune system comply. The first and most obvious one is to use multiple vaccine doses over an extended period of time. The next is to use adjuvants- which is by no means exclusive to the neonate. However, the most important solution is probably maternal immunization- immunizing the mother while pregnant so her IgG can be transferred through the placenta and help protect the infant for the first few months of life. This is particularly important for protection for pathogens requiring humoral immunity, but by no means unimportant for other pathogens. Additionally, there is some evidence that certain vaccines (in particular BCG) can induce a state of nonspecific “trained immunity” where the innate immune system mediates memory-like responses to multiple antigens at once. There is however the issue of maternal antibody interference, in which the antibodies from the mother inhibit effective vaccine responses by the infant.
On the other side of things is the challenge of an elderly immune system. Elderly people undergo a set of changes broadly known as immunosenescence. The adaptive immune system relies on the ability to rapidly expand the number of cells it needs to deal with an infection, particularly the cells of the adaptive immune system, which is known as clonal expansion. The issue however, is that all cells have a Hayflick limit. Cells will only divide a limited number of times before committing suicide (this is largely thought to be a protective mechanism against cancer. As you age, you accumulate DNA damage in your cells that can lead to the formation of tumors or malignancies- hence age is the biggest risk factor in most cancers). Naturally, the older you are, the more divisions each of your cells have undergone. Thus rapid expansion of specific cell populations is more difficult. This is thought to be a major contributor to the resurgence of shingles and other herpesviral infections in the elderly. In addition, this is further complicated by the fact that the thymus undergoes a process called involution- basically it withers away into nothing- once you hit about puberty. The thymus is the site of T cell synthesis. The mechanisms are reviewed here, but in essence this manages to promote a constant low grade state of inflammation (which is known as- yes really- inflammaging) and also reduced capacity to respond to new antigens. T cells can also be made outside the thymus, however, which is reviewed here. Inflammaging results in a chronic, low grade sterile inflammatory state from the stimulation of the innate immune system (in essence it triggers a negative feedback loop of anti-inflammatory mechanisms which make it harder to initiate immune responses). I The trouble is that this essentially sets the baseline for an adaptive immune response to occur even higher, and thus elderly immune systems may require some coaxing. Inflammaging is probably a post unto itself so I leave readers the linked review for further information. Elderly people also struggle with B cell responses and the generation of high affinity antibodies.
Source: Figure 38.3 Rich R. Clinical immunology. 5th ed. Elsevier; 2019.
Source: Siegrist, C.-A., & Aspinall, R. (2009). B-cell responses to vaccination at the extremes of age. Nature Reviews Immunology, 9(3), 185–194. doi:10.1038/nri2508
Strategies to deal with immunosenescence. Source: Rich R. Clinical immunology. 5th ed. Elsevier; 2019.
As I have written previously, killed vaccines contain all their PAMPs (pathogen-associated molecular patterns) and thus do not require adjuvants to work. However, there is an exception: influenza vaccines for the elderly are adjuvanted with MF59- an oil-in-water emulsion comprising squalene, Tween-80, and Span 85 that serves to recruit the innate immune system and promote pro-inflammatory cytokine production. Alternatively, there is a high-dose vaccine for the elderly aiming to overcome the challenges of immunosenescence. The challenges here, however, are a bit more complex to deal with than for with neonates and require further research. A non-comprehensive summary of the challenges with vaccinology at the extremes of age is in the table below:
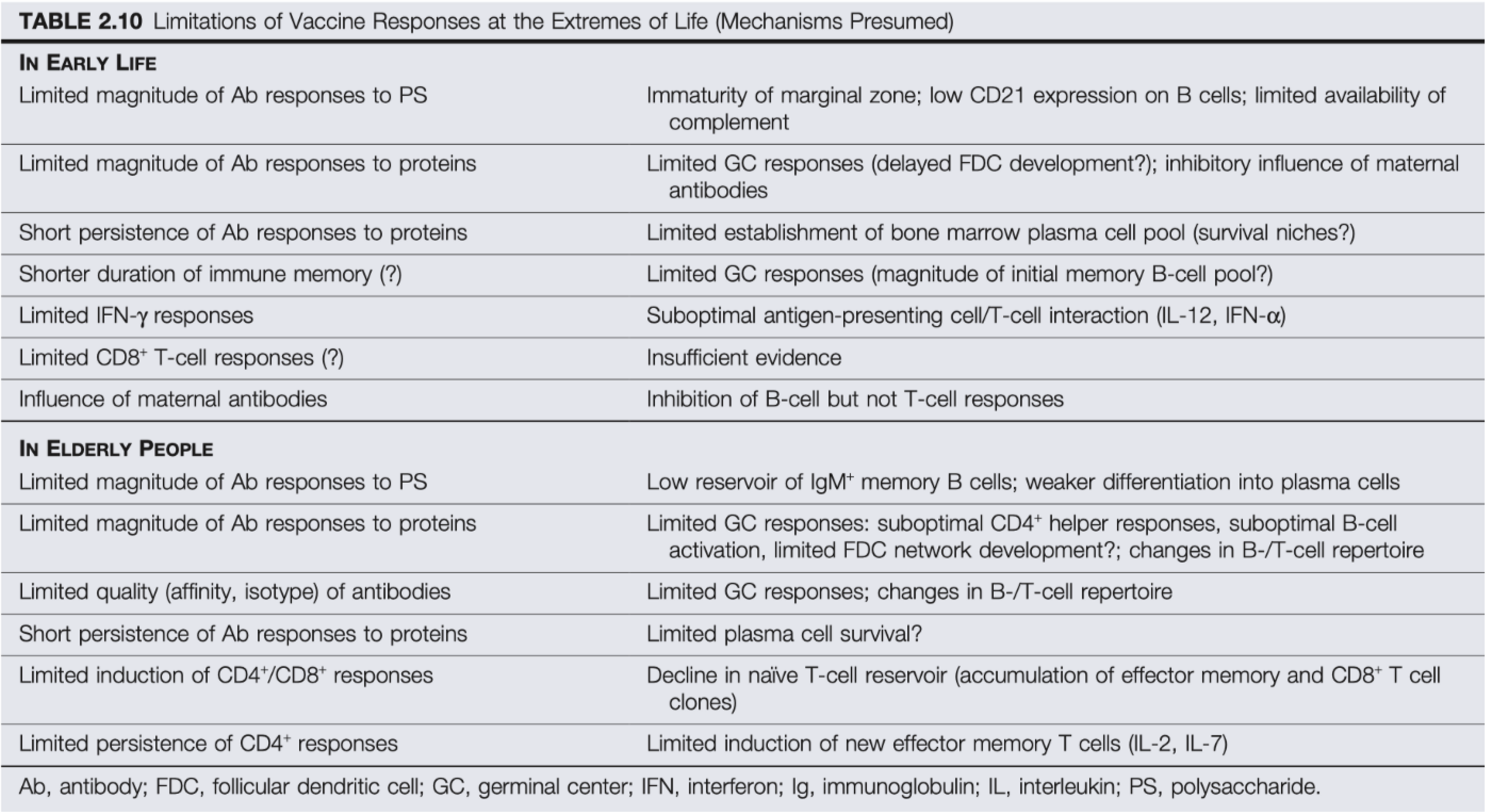
Source: Table 2.10 Plotkin’s Vaccines 7th Edition
Sterilizing Immunity
Some people have raised the concern that a vaccine candidate for COVID-19 should provide sterilizing immunity. This is a complex one. The reality is that most vaccines do not work in the manner that they are generally perceived to, which is known as sterilizing immunity. Sterilizing in this contest refers to cleanliness- not fertility. This is where the immune system complete eliminates the pathogen before it can establish an infection e.g. every single virion in an inoculum gets neutralized by an antibody before a single one can enter a cell. However, most vaccines do not induce sterilizing immunity, and furthermore, they do not need to for effective disease control. It is generally common for vaccines to induce a state of protective immunity in which the recipient, should they encounter the pathogen, to experience a subclinical infection (one which does not cause symptoms). This is sufficient for control of a disease on a public health level and a personal health level. For example, Hepatitis B vaccines are extremely effective at preventing disease (80–100% after 2 doses) and contain the surface- but not the core- antigen of the virus. Healthcare workers who receive the vaccine however demonstrate antibodies to the core antigen as well- which indicates that they have been exposed to hepatitis B and it was able to establish some level of infection- but the healthcare workers in question did not develop hepatitis B, and could not spread hepatitis B. Even if the vaccine is a leaky one (unable to effectively prevent disease but can reduce severity) that would be worthwhile. Sterilizing immunity however may be required for control of some chronic viral infections e.g. hepatitis C.
Induction of Mucosal Immunity
Source: Janeway’s Immunobiology 9th Edition Figure 12.1
The functional separation of the mucosal immune system from the systemic (circulatory) compartment means that different vaccination approaches must be employed to achieve effective mucosal immunity.
Chapter 55 from Mestecky J, Strober W, Russell M, Cheroutre H, Lambrecht B, Kelsall B. Mucosal Immunology. Burlington: Elsevier Science; 2015.
Inducing effective responses at mucosal surfaces (see above for what body parts fall under this category) is a more complex task than it may sound at first blush. In a topological sense, your mucosa lie on the outside of your body, compared with, for instance, your blood vessels and internal organs- which makes it a challenge because immune responses arising from the circulatory system cannot readily access mucosal surfaces. Consider for instance the oral polio vaccine (OPV) and the inactivated polio vaccine (OVP). OPV is associated with vaccine-associated paralytic polio (VAPP) in about 1 per 2.7 million doses. IPV on the other hand, is not. So, why use OPV instead? There are several reasons, but a major one is that OPV, but not IPV, induces effective mucosal immune responses that are necessary to interrupt transmission of the virus. The systemic immune system represents a separate compartment in the body for immunity compared with the mucosal immune system- and furthermore the bulk of immunocytes (the cells of the immune system) lie in the mucosal immune system. Mucosal immunity generally requires the use of live vaccines, or requires unique adjuvants to induce outside the context of live vaccines. Generally mucosal routes of administration are preferred however because they don’t require special training to administer and can be used to induce specific responses at that surface, typically where the pathogen might ordinarily be encountered e.g. poliovirus spreads by the fecal-oral route and so drinking the vaccine makes transmission through a population much more difficult, and also does not require someone trained in giving subcutaneous injections.
There are, however, a number of challenges. Consider for instance your diet- every single day you take food in through your gastrointestinal tract. The food you eat is rife with antigens that could potentially induce immune responses and lead to tissue damage (and they sometimes do- as in the case of celiac disease or food allergies). However, broadly speaking, mechanisms of mucosal tolerance are present to ensure a state of non-responsiveness from the mucosal immune system. Under physiological conditions, this is quite important- consider how hard it becomes to do basic things when mucosal tissues are inflamed (one of the hallmarks of inflammation is loss of function) e.g. maintaining good gas exchange in the lungs while afflicted with pneumonia. However, this is problematic for vaccination as it requires workarounds as we do indeed want an effective immune response at the mucosal surfaces. One approach is the use of mucosal adjuvants- but issues arise here too. For instance, cholera toxin is noted to be an excellent mucosal adjuvant- but cholera toxin is still… a toxin that can be quite dangerous even in small quantities and thus for effective adjuvant use may require targeted mutation to inactivate. Additionally, a major effector of mucosal immune responses is the secretory IgA molecule- but the persistence of these IgA molecules is difficult to achieve and often seems to require that the antigen it is matched against is present at the mucosal site. We do make plasma cells (highly specialized B cells) that produce large quantities of IgA in response to vaccinations and infection- but as these are confined to the circulatory compartment, they cannot readily access mucosal sites for protection.
Primary and Secondary Vaccine Failure
In a basic sense, there are two ways a vaccine can fail, which are known as primary and secondary vaccine failure. In primary vaccine failure, there is a failure of the host’s immune system to respond to the vaccine and thus antibodies (or the pertinent effector for that vaccine) are never made and thus the host never gains any protection. Secondary vaccine failure refers to the waning of immunity over time to sub-protective levels after an initial response.
Primary vaccine failure occurs based on both host and vaccine factors. In the case of vaccine factors, this could be using the incorrect route of administration, issues with proper attenuation of the vaccine antigens, and issues with storage of the vaccine prior to administration. The good news is that these factors are readily modifiable (though of course the challenges of logistic hurdles should not be discounted). Host factors on the other hand are far more complex. For routinely administered vaccines, 2–10% of healthy vaccinees are non-responders, and this is especially well documented with hepatitis B. To date a number of risk factors have been described including obesity, renal disease, heavy smoking or drinking, age (as discussed above) and variations in HLA (human leukocyte antigen- the MHC proteins) proteins. Some mechanistic explanation comes from the observation that non-responders seem to have higher levels of anti-inflammatory cell populations e.g. B regulatory cells.
Thus far, workarounds for primary vaccine failure have been challenging. An obvious solution is to give more frequent doses of the vaccines (and this would readily improve issues with secondary vaccine failure) or increase the dose of antigens. However, more work needs to be done to satisfactorily address this. There is some good news however, in that herd immunity means that with sufficient vaccine uptake and sufficiently good efficacy, primary and secondary vaccine failure do not guarantee disease in non-responders.
The Importance of Good Animal Models
There are substantial ethical constraints on the use of humans in clinical studies, as I have written about previously. Hence, prior to initiating studies in humans, vaccines go through extensive pre-clinical testing. As discussed earlier, designing a good vaccine often requires some fairly extensive knowledge of the immune system’s response to the infection and how it would ordinarily clear it. In the context of an animal model, this typically requires sacrificing the subjects (which I think I don’t have to explain, is not something you can do with humans). Research in HIV has been held back a great deal because the virus has a very narrow host range. The virus appears to have come from the chimpanzee virus SIVcpz- but infection of chimpanzees with HIV does not seemingly cause disease. Macaque species are therefore commonly used in AIDS research, but the use of macaques and other nonhuman primates creates substantial ethical quandaries in research, elaborated upon here. I don’t know of anyone who likes animal testing- but thus far we don’t have the science to be able to skip it altogether. Improvements in biotechnology however have done a great deal to enhance animal models, in particular systems like CRISPR-Cas and Cre-LOX. To be able to study a virus requires cells that are both susceptible (they express the viral receptor) and permissive (they have the metabolic capacity to support viral replication). A major reason viruses are often restricted to a single species is because the receptor for the virus is relatively unique to that species to a degree that makes it impossible for the virus to invade other species. We can insert transgenes that would render the cells of an organism susceptible. Additionally, humanized mice have recently gained ground as a model organism. These are mice which undergo genetic modification to have an immune system that is more human-like, which is important because there are substantial differences between mice and human immune systems.
Table 1 from Akkina, R. (2013). Human immune responses and potential for vaccine assessment in humanized mice. Current Opinion in Immunology, 25(3), 403–409. doi:10.1016/j.coi.2013.03.009
Another animal model that is becoming increasingly important for studying these diseases is the bat. Bats are viral reservoirs and have some unique immunological adaptations which manage to keep them from becoming ill. There are a few explanations proposed but it appears that uniqueness of the bat’s immune system- serving as hosts for these viruses without becoming sickened themselves- arose as a consequence of their evolution of flight. In the process of flying, bats require a great deal of energy and in the process generate reactive oxygen species (ROS) which are oxygen-containing molecules that readily react with a number of biological molecules. ROS can cause tissue damage (and also play key roles in host defense). To control the resultant inflammation, bats appear to lack PYHIN domain-containing proteins which disrupts their ability to generate an inflammasome (see my post on aluminum-based adjuvants for the significance of that). While fascinating and important, bats are very difficult animal models for research for a number of reasons. For one thing, there are relatively few bat cell lines. Additionally, bats as a group have tremendous genotypic and phenotypic variation.
Immunodominance
Source: Mathew, N. R., & Angeletti, D. (2020). Recombinant Influenza Vaccines: Saviors to Overcome Immunodominance. Frontiers in Immunology, 10. doi:10.3389/fimmu.2019.02997
With viruses which are antigenically variable (meaning not SARS-CoV-2)- like influenza and HIV- we encounter the issue of immunodominance. Immunodominance is a term that describes the tendency for the immune system to respond to a particular epitope (a region on an antigen that gets recognized) over other epitopes. This turns out to be a huge problem for making effective vaccines against influenza and HIV. In the case of influenza, the hemagglutinin (H) protein is used to help facilitate invasion of cells, and thus it seems logical that the immune system should target it to control infection. However, the H protein has a stalk (also called a stem) and a head- and the head is highly variable across strains while the stalk is relatively conserved. Unfortunately, epitopes on the head of the protein are immunodominant, which makes cross-protection across many strains of influenza difficult to attain. What is curious however is that anti-stem targeting B cells are common in patients who have influenza infection or vaccinations- but they are outcompeted by B cells which make antibodies against the head. Thus, one proposed solution is to exclude the head domain of the H protein altogether in vaccines and instead use just the stem (some others are described in the figure below).
Considerations of immunodominance do also apply to T cells, however (and in fact the term came about from observing how T cells respond to different epitopes, rather than B cells). As I discussed earlier, when antigen appears inside the body, it gets taken up by professional antigen presenting cells (pAPCs) which process the discrete epitopes and display them on an MHC protein for T cells, and in this case, MHC class II because it is exogenous antigen. I won’t discuss the mechanisms here, but there is a phenomenon known as epitope accessibility which says that the epitope that best fits inside the MHC protein groove is the one that will be presented- not unlike Cinderella with her glass slipper.
Immunodominance is not an inherent deficiency in how our immune system works and in fact can sometimes be to our benefit. The immunodominant epitopes on measles H and F proteins for example lie on the regions required for the virus to be able to bind and invade cells, making it relatively easy to produce neutralizing antibodies. Furthermore, there is some evidence that immunodominance may contribute to our immune system’s tolerance of self-antigen.
Source: Figure 4 from Ruckwardt, T. J., Morabito, K. M., & Graham, B. S. (2019). Immunological Lessons from Respiratory Syncytial Virus Vaccine Development. Immunity, 51(3), 429–442. doi:10.1016/j.immuni.2019.08.007
Vaccine Immunopathology
Though vaccine failure as described above is potentially serious, vaccine-induced immunopathology is even more so and has been noted with prior attempts to make vaccines against SARS-CoV-1, RSV, Dengue, and measles. This is best characterized in the case of RSV so I will focus on it here.
RSV is a devastating respiratory infection at the extremes of age, in particular in the very young but also in the elderly. In fact, Feigin and Cherry’s Textbook of Pediatric Infectious Disease goes as far as to say:
Respiratory syncytial virus (RSV) is the most important respiratory pathogen of infancy and early childhood and the major cause of hospitalization for bronchiolitis and pneumonia in infants globally. (39,101–103,152,225,228) In the United States, RSV has been estimated to be the leading cause of all hospitalizations for infants, resulting in an estimated 74,000 to 126,000 hospitalizations each year from 1994 to 1996. (175,250) More recent studies suggest that these figures are lower than the current number of infant hospitalizations associated with RSV infections and that the associated annual costs are approximately $2.6 billion. (131,133,145) Furthermore, the considerable burden of RSV infections among older children, adults, and outpatients has not been recognized.
Summary of antibody-dependent enhancement. Source: Rothman, A. L. (2011). Immunity to dengue virus: a tale of original antigenic sin and tropical cytokine storms. Nature Reviews Immunology, 11(8), 532–543. doi:10.1038/nri3014
We have been attempting a vaccine against RSV since the 1960s, but have yet to be successful. The major reason appears to be the resulting vaccine-enhanced disease that occurs with vaccine candidates. In 1967, a vaccine candidate used a formalin-inactivated virus to vaccinate children. When the children encountered the virus, they were not only not protected from the disease, but actually had more severe disease, with two children in the trial even having died. Needless to say, the vaccine candidate was abandoned. The underlying cause of the worse outcomes appears to have been a hyperinflammatory state mediated by Th2 cells. The Th2 cells activated and recruited eosinophils- white blood cells which are key in the control of parasitic infections but are behind much of the pathology in allergic disease including anaphylaxis- which resulted in bronchopneumonia and lung injury. There is still much about why this happened that is unclear and not entirely explainable with our current understanding of immunology. For instance, while eosinophils are typically regarded as being inherent to Th2 immune responses, Hotez, Bottazzi, and Corry discuss that viral vectored vaccines can seemingly give rise to Th17-mediated immunopathology which also involves eosinophils- which may be a potential concern for viral vector candidates of SARS-CoV-2 vaccines (this may help to explain Dengvaxia associated immunopathology as well). Importantly, vaccine enhanced disease does not arise de novo- the reason that a hyper-Th2 response occurs is that, despite being a virus, RSV has antigens that shift the immune system away from more protective Th1-like responses and towards Th2 responses. The exact role of the immune system and mechanism of enhanced disease is unclear, however. A more detailed review of the host-pathogen interactions of RSV with the immune system and how immunopathology arises can be found here.
The situation is not, however, hopeless. A recent study describes that through the use of CpG and monophosphoryl lipid A antigens, a Th2-driven immune response against RSV can be suppressed and a protective Th1-dominant one can be instituted. However, note that this is a mouse study and thus has likely decades to go before attaining fulfillment at the bedside. At the very least, however, the proof of concept suggests that the chance for a safe and effective RSV vaccine does exist and we should continue to pursue it. I should also point out that since the debacle with the formalin-inactivated RSV vaccine, the FDA screens candidates for vaccines against respiratory viruses especially stringently to rule out immune enhancement. However, it is still important to realize that this does not appear to be an issue for many respiratory viruses e.g. influenza.
You may have heard in the news another form known as antibody-dependent enhancement (ADE). This is also not completely understood and seems to be an issue for situations in which closely related but distinct pathogens infect a host. Memory responses will always outcompete primary immune responses, so the concept is that after an initial exposure to a closely related pathogen, the immune system is primed to produce a given kind of antibody that will be neutralizing for that particular pathogen. However, in a subsequent encounter with a closely related pathogen, those same antibodies will be recalled, but will not be neutralizing (sometimes called binding antibodies). The result is that the antibodies end up facilitating the spread of the virus throughout the body and result in more severe disease. This is best documented with Dengue but has also been observed in animal models of SARS infection. In general, antibody dependent enhancement is a very rare phenomenon and antibodies are protective in infectious disease rather than pathogenic.
Fortunately, there are workarounds for antibody-dependent enhancement. One approach is to ensure that all the antibodies will be neutralizing by including only highly conserved epitopes in the vaccine that will be constant from strain to strain. In particular, if the epitope has a functional necessity for infection, this approach is an elegant solution e.g. using only the receptor-binding domain of SARS-CoV-2’s spike protein. Note of course that this approach does require vaccine adjuvants.
Conclusions
The production of effective vaccines faces numerous challenges and generally requires considerable time before the issues are surmounted by research efforts.
References
Angeletti, D., & Yewdell, J. W. (2018). Understanding and Manipulating Viral Immunity: Antibody Immunodominance Enters Center Stage. Trends in Immunology, 39(7), 549–561. doi:10.1016/j.it.2018.04.008
Mathew, N. R., & Angeletti, D. (2020). Recombinant Influenza Vaccines: Saviors to Overcome Immunodominance. Frontiers in Immunology, 10. doi:10.3389/fimmu.2019.02997
Kim, A., & Sadegh-Nasseri, S. (2015). Determinants of immunodominance for CD4 T cells. Current Opinion in Immunology, 34, 9–15. doi:10.1016/j.coi.2014.12.005
Akram, A., & Inman, R. D. (2012). Immunodominance: A pivotal principle in host response to viral infections. Clinical Immunology, 143(2), 99–115. doi:10.1016/j.clim.2012.01.015
Akkina, R. (2013). Human immune responses and potential for vaccine assessment in humanized mice. Current Opinion in Immunology, 25(3), 403–409. doi:10.1016/j.coi.2013.03.009
Kumar, P., Chen, K., & Kolls, J. K. (2013). Th17 cell based vaccines in mucosal immunity. Current Opinion in Immunology, 25(3), 373–380. doi:10.1016/j.coi.2013.03.011
Angeletti, D., & Yewdell, J. W. (2017). Is It Possible to Develop a “Universal” Influenza Virus Vaccine? Cold Spring Harbor Perspectives in Biology, 10(7), a028852. doi:10.1101/cshperspect.a028852
Ruckwardt, T. J., Morabito, K. M., & Graham, B. S. (2019). Immunological Lessons from Respiratory Syncytial Virus Vaccine Development. Immunity, 51(3), 429–442. doi:10.1016/j.immuni.2019.08.007
Kollmann T, Marchant A, Way S. Vaccination strategies to enhance immunity in neonates. Science. 2020;368(6491):612–615.
Mestecky J, Strober W, Russell M, Cheroutre H, Lambrecht B, Kelsall B. Mucosal Immunology. Burlington: Elsevier Science; 2015.
Rich R. et al. Clinical Immunology. 5th ed. Elsevier; 2019.
Wood, N., & Siegrist, C.-A. (2011). Neonatal immunization: where do we stand? Current Opinion in Infectious Diseases, 24(3), 190–195. doi:10.1097/qco.0b013e328345d563
Kurts, C., Robinson, B. W. S., & Knolle, P. A. (2010). Cross-priming in health and disease. Nature Reviews Immunology, 10(6), 403–414. doi:10.1038/nri2780
Pardi, N., Hogan, M. J., Porter, F. W., & Weissman, D. (2018). mRNA vaccines — a new era in vaccinology. Nature Reviews Drug Discovery, 17(4), 261–279. doi:10.1038/nrd.2017.243
Pardi, N., Hogan, M. J., & Weissman, D. (2020). Recent advances in mRNA vaccine technology. Current Opinion in Immunology, 65, 14–20. doi:10.1016/j.coi.2020.01.008
Zhang C, Maruggi G, Shan H, Li J. Advances in mRNA Vaccines for Infectious Diseases. Frontiers in Immunology. 2019;10.
Thomas, R., Wang, W., & Su, D.-M. (2020). Contributions of Age-Related Thymic Involution to Immunosenescence and Inflammaging. Immunity & Ageing, 17(1). doi:10.1186/s12979–020–0173–8
Rocha, B., Vassalli, P., & Guy-Grand, D. (1992). The extrathymic T-cell development pathway. Immunology Today, 13(11), 449–454. doi:10.1016/0167–5699(92)90074-h
Mosca, F., Tritto, E., Muzzi, A., Monaci, E., Bagnoli, F., Iavarone, C., … De Gregorio, E. (2008). Molecular and cellular signatures of human vaccine adjuvants. Proceedings of the National Academy of Sciences, 105(30), 10501–10506. doi:10.1073/pnas.0804699105
Siegrist, C.-A., & Aspinall, R. (2009). B-cell responses to vaccination at the extremes of age. Nature Reviews Immunology, 9(3), 185–194. doi:10.1038/nri2508
Mathew, N. R., & Angeletti, D. (2020). Recombinant Influenza Vaccines: Saviors to Overcome Immunodominance. Frontiers in Immunology, 10. doi:10.3389/fimmu.2019.02997
Holmgren, J., & Czerkinsky, C. (2005). Mucosal immunity and vaccines. Nature Medicine, 11(4s), S45–S53. doi:10.1038/nm1213
Chawla, B., Mahajan, B., Oakley, M., Majam, V. F., Belmonte, A., Sedegah, M., … Kumar, S. (2019). Antibody-dependent IFN-γ-independent sterilizing immunity, induced by a subunit malaria vaccine. Infection and Immunity. doi:10.1128/iai.00236–19
Joffre, O. P., Segura, E., Savina, A., & Amigorena, S. (2012). Cross-presentation by dendritic cells. Nature Reviews Immunology, 12(8), 557–569. doi:10.1038/nri3254
Yewdell, J. W. (2006). Confronting Complexity: Real-World Immunodominance in Antiviral CD8+ T Cell Responses. Immunity, 25(4), 533–543. doi:10.1016/j.immuni.2006.09.005
Angeletti, D., Kosik, I., Santos, J. J. S., Yewdell, W. T., Boudreau, C. M., Mallajosyula, V. V. A., … Yewdell, J. W. (2019). Outflanking immunodominance to target subdominant broadly neutralizing epitopes. Proceedings of the National Academy of Sciences, 201816300. doi:10.1073/pnas.1816300116
Lin, Q., Zhu, L., Ni, Z., Meng, H., & You, L. (2020). Duration of serum neutralizing antibodies for SARS-CoV-2: Lessons from SARS-CoV infection. Journal of Microbiology, Immunology and Infection. doi:10.1016/j.jmii.2020.03.015
Siegrist, C.-A., & Aspinall, R. (2009). B-cell responses to vaccination at the extremes of age. Nature Reviews Immunology, 9(3), 185–194. doi:10.1038/nri2508
Sahin, U., Karikó, K., & Türeci, Ö. (2014). mRNA-based therapeutics — developing a new class of drugs. Nature Reviews Drug Discovery, 13(10), 759–780. doi:10.1038/nrd4278
Gustafson, C. E., Kim, C., Weyand, C. M., & Goronzy, J. J. (2020). Influence of immune aging on vaccine responses. Journal of Allergy and Clinical Immunology, 145(5), 1309–1321. doi:10.1016/j.jaci.2020.03.017
Mestas, J., & Hughes, C. C. W. (2004). Of Mice and Not Men: Differences between Mouse and Human Immunology. The Journal of Immunology, 172(5), 2731–2738. doi:10.4049/jimmunol.172.5.2731
Hatziioannou, T., & Evans, D. T. (2012). Animal models for HIV/AIDS research. Nature Reviews Microbiology, 10(12), 852–867. doi:10.1038/nrmicro2911
Schountz, T. (2014). Immunology of Bats and Their Viruses: Challenges and Opportunities. Viruses, 6(12), 4880–4901. doi:10.3390/v6124880
Calisher, C. H., Childs, J. E., Field, H. E., Holmes, K. V., & Schountz, T. (2006). Bats: Important Reservoir Hosts of Emerging Viruses. Clinical Microbiology Reviews, 19(3), 531–545. doi:10.1128/cmr.00017–06
Walker, R. L. (2018). Virtue, Vice, and “Voracious” Science: How should we approach the ethics of primate research? Perspectives in Biology and Medicine, 61(1), 130–146. doi:10.1353/pbm.2018.0032
DeGrazia, D. (2016). Nonhuman Primates, Human Need, and Ethical Constraints. Hastings Center Report, 46(4), 27–28. doi:10.1002/hast.601
Plotkin S, Orenstein W, Offit P, Edwards K. Plotkin’s Vaccines . 7th ed. Saint Louis: Elsevier; 2017.
Murphy K, Weaver C. Janeway’s Immunobiology. 9th ed.; 2017
Ursula Wiedermann, Erika Garner-Spitzer & Angelika Wagner (2016) Primary vaccine failure to routine vaccines: Why and what to do?, Human Vaccines & Immunotherapeutics, 12:1, 239–243, DOI: 10.1080/21645515.2015.1093263
Orr, M. T., Beebe, E. A., Hudson, T. E., Moon, J. J., Fox, C. B., Reed, S. G., & Coler, R. N. (2014). A Dual TLR Agonist Adjuvant Enhances the Immunogenicity and Protective Efficacy of the Tuberculosis Vaccine Antigen ID93. PLoS ONE, 9(1), e83884. doi:10.1371/journal.pone.0083884
Horlock C. Production of MHC Class I Tetramers | British Society for Immunology. Immunology.org. 2020 https://www.immunology.org/public-information/bitesized-immunology/experimental-techniques/production-mhc-class-i-tetramers
Ho, N. I., Huis in ’t Veld, L. G. M., Raaijmakers, T. K., & Adema, G. J. (2018). Adjuvants Enhancing Cross-Presentation by Dendritic Cells: The Key to More Effective Vaccines? Frontiers in Immunology, 9. doi:10.3389/fimmu.2018.02874
Blander, J. M. (2018). Regulation of the Cell Biology of Antigen Cross-Presentation. Annual Review of Immunology, 36(1), 717–753. doi:10.1146/annurev-immunol-041015–055523
MacLeod, M. K. L., McKee, A. S., David, A., Wang, J., Mason, R., Kappler, J. W., & Marrack, P. (2011). Vaccine adjuvants aluminum and monophosphoryl lipid A provide distinct signals to generate protective cytotoxic memory CD8 T cells. Proceedings of the National Academy of Sciences, 108(19), 7914–7919. doi:10.1073/pnas.1104588108
Rogers GL, Shirley JL, Zolotukhin I, Kumar SRP, Sherman A, et al. 2017. Plasmacytoid and conventional dendritic cells cooperate in crosspriming AAV capsid-specific CD8 + T cells. Blood 129:3184–95
Witkowski, J. M., Larbi, A., Le Page, A., & Fülöp, T. (2020). Natural Killer Cells, Aging, and Vaccination. Interdisciplinary Topics in Gerontology and Geriatrics, 18–35. doi:10.1159/000504493
Norbury, C. C. (2016). Defining cross presentation for a wider audience. Current Opinion in Immunology, 40, 110–116. doi:10.1016/j.coi.2016.04.003
Cruz, F. M., Colbert, J. D., Merino, E., Kriegsman, B. A., & Rock, K. L. (2017). The Biology and Underlying Mechanisms of Cross-Presentation of Exogenous Antigens on MHC-I Molecules. Annual Review of Immunology, 35(1), 149–176. doi:10.1146/annurev-immunol-041015–055254
Broadly Neutralizing Antibodies. IAVI. 2020. iavi.org/our-science/broadly-neutralizing-antibodies
There is one, and only one strain of SARS-CoV-2. Virology.ws. 2020. https://www.virology.ws/2020/05/07/there-is-one-and-only-one-strain-of-sars-cov-2/
Bottlenecks and founder effects. Evolution.berkeley.edu. 2020.
Niewiesk, S. (2014). Maternal Antibodies: Clinical Significance, Mechanism of Interference with Immune Responses, and Possible Vaccination Strategies. Frontiers in Immunology, 5. doi:10.3389/fimmu.2014.00446
Shay, J. W., & Wright, W. E. (2000). Hayflick, his limit and cellular ageing. Nature Reviews Molecular Cell Biology, 1(1), 72–76. doi:10.1038/35036093
Muralidharan, A., Li, C., Wang, L., & Li, X. (2016). Immunopathogenesis associated with formaldehyde-inactivated RSV vaccine in preclinical and clinical studies. Expert Review of Vaccines, 16(4), 351–360. doi:10.1080/14760584.2017.1260452
Acosta, P. L., Caballero, M. T., & Polack, F. P. (2015). Brief History and Characterization of Enhanced Respiratory Syncytial Virus Disease. Clinical and Vaccine Immunology, 23(3), 189–195. doi:10.1128/cvi.00609–15
Smatti, M. K., Al Thani, A. A., & Yassine, H. M. (2018). Viral-Induced Enhanced Disease Illness. Frontiers in Microbiology, 9. doi:10.3389/fmicb.2018.02991
Lee, Y., Ko, E.-J., Kim, K.-H., Lee, Y.-T., Hwang, H. S., Kwon, Y.-M., … Kang, S. M. (2019). A unique combination adjuvant modulates immune responses preventing vaccine-enhanced pulmonary histopathology after a single dose vaccination with fusion protein and challenge with respiratory syncytial virus. Virology, 534, 1–13. doi:10.1016/j.virol.2019.05.010
Feigin R, Cherry J, Harrison G. Textbook of Pediatric Infectious Diseases. 8th ed. Philadelphia: Elsevier Health Sciences; 2019.
Pepper, M., & Jenkins, M. K. (2011). Origins of CD4+ effector and central memory T cells. Nature Immunology, 12(6), 467–471. doi:10.1038/ni.2038
Hotez P, Bottazzi M, Corry D. The potential role of Th17 immune responses in coronavirus immunopathology and vaccine-induced immune enhancement. Microbes and Infection. 2020.
Mangodt, T. C., Van Herck, M. A., Nullens, S., Ramet, J., De Dooy, J. J., Jorens, P. G., & De Winter, B. Y. (2015). The role of Th17 and Treg responses in the pathogenesis of RSV infection. Pediatric Research, 78(5), 483–491. doi:10.1038/pr.2015.143
Kam, Y. W., Kien, F., Roberts, A., Cheung, Y. C., Lamirande, E. W., Vogel, L., … Altmeyer, R. (2007). Antibodies against trimeric S glycoprotein protect hamsters against SARS-CoV challenge despite their capacity to mediate FcγRII-dependent entry into B cells in vitro. Vaccine, 25(4), 729–740. doi:10.1016/j.vaccine.2006.08.011
Jiang S, He Y, Liu S. SARS Vaccine Development. Emerging Infectious Diseases. 2005;11(7):1016–1020.
Halstead, SB. Dengue Antibody-Dependent Enhancement: Knowns and Unknowns. (n.d.). Antibodies for Infectious Diseases, 249–271. doi:10.1128/microbiolspec.aid-0022–2014
Rothman, A. L. (2011). Immunity to dengue virus: a tale of original antigenic sin and tropical cytokine storms. Nature Reviews Immunology, 11(8), 532–543. doi:10.1038/nri3014
Chen, W.-H., Hotez, P. J., & Bottazzi, M. E. (2020). Potential for developing a SARS-CoV receptor-binding domain (RBD) recombinant protein as a heterologous human vaccine against coronavirus infectious disease (COVID)-19. Human Vaccines & Immunotherapeutics, 1–4. doi:10.1080/21645515.2020.1740560
Doores, K. J. (2015). The HIV glycan shield as a target for broadly neutralizing antibodies. FEBS Journal, 282(24), 4679–4691. doi:10.1111/febs.13530